
Molecular Dynamics
Graph Neural Network for Prediction of Molecular Properties
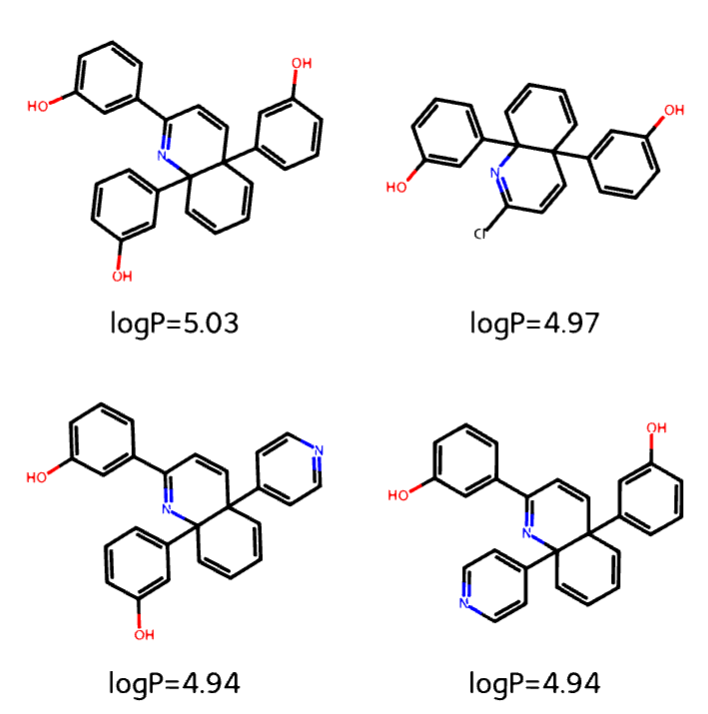
● Designed and implemented a full molecular generation and optimization pipeline using RDKit and PyTorch Geometric,
programmatically enumerating and sampling Quinoline derivatives using node-edge tensors across multi-site
functionalization spaces (>50,000 Quinoline samples used for training)
● Developed a Graph Neural Network (GNN) operating directly on molecular graphs, encoding physicochemical features
to predict LogP with end-to-end differentiable learning
● Applied GNN to >50 billion molecular combinations, ranking synthetically feasible candidates based on predicted logP
Sintering of Nanoparticles into a Polycrystalline Solid
● This molecular dynamics simulation models the thermal sintering of metallic nanoparticles using LAMMPS. Initially, separate particles gradually coalesce under elevated temperatures. The process captures grain boundary formation at the nanoscale level. Simulation done using LAMMPS, visualization using OVITO, VMD, and Blender 3D.

Molecular Dynamics Application
● Architected and implemented a modular Python application for physics-based molecular dynamics simulation
orchestration, abstracting low-level command prompt configuration and file management into vastly improved
programmatic workflows with much greater accessibility
● Developed AI-enhanced automation pipelines for simulation setup, including data-driven selection and optimization of
simulation parameters, timestep controls, and LAMMPS run configurations
● Integrated GPU-accelerated computing via CUDA to enable scalable, high-throughput molecular simulations and
post-processing of atomistic trajectory data
Thermal Denaturation of GB1 Protein in Water
● This GROMACS simulation models the denaturation of the 1PGA protein (Protein G B1 domain) under high-temperature conditions in a water environment. As the temperature increases, the protein’s secondary and tertiary structures destabilize, leading to unfolding and loss of native conformation. The simulation captures the dynamic process of thermal denaturation, illustrating how heat disrupts intermolecular forces between the amino acid subunits of the protein. Simulation utilizes GROMACS for simulations, with OVITO, VMD, and Blender 3D for visualizations.

Tensile Fracture of a Crystalline Silicon Lattice
● This LAMMPS simulation demonstrates the mechanical failure of a crystalline silicon lattice under uniaxial tensile stress. As the material is stretched, atomic bonds begin to break, leading to crack initiation and propagation. The simulation visualizes the fracture process at the atomic scale. Visualization done using OVITO, VMD, and Blender 3D.

